Arlequin ver 3.11
(released 19 February 2007)
|
Arlequin ver 3.11
|
|
|
|
Arlequin is the French translation of "Arlecchino", a famous character of the Italian "Commedia dell'Arte". As a character he has many aspects, but he has the ability to switch among them very easily according to its needs and to necessities. This polymorphic ability is symbolized by his colorful costume, from which the Arlequin icon was designed.
The goal of Arlequin is to provide the average user in population genetics with quite a large set of basic methods and statistical tests, in order to extract information on genetic and demographic features of a collection of population samples.
The graphical interface is designed to allow users to rapidly select the different analyses they want to perform on their data. We felt important to be able to explore the data, to analyze several times the same data set from different perspectives, with different selected options.
The statistical tests implemented in Arlequin have been chosen such as to minimize hidden assumptions and to be as powerful as possible. Thus, they often take the form of either permutation tests or exact tests, with some exceptions.
Finally, we wanted Arlequin to be able to handle genetic data under many different forms, and to try to carry out the same types of analyses irrespective of the format of the data.
Because Arlequin has a rich set of features and many options, it means that the user has to spend some time in learning them. However, we hope that the learning curve will not be that steep.
Arlequin is made available free of charge, as long as we have enough local resources to support the development of the program.
The analyses Arlequin can perform on the data fall into two main categories: intra-population and inter-population methods. In the first category statistical information is extracted independently from each population, whereas in the second category, samples are compared to each other.
|
Intra-population methods: |
Short description: |
|
Standard indices |
Some diversity measures like the number of polymorphic sites, gene diversity. |
|
Molecular diversity |
Calculates several diversity indices like nucleotide diversity, different estimators of the population parameter q. |
|
Mismatch distribution |
The distribution of the number of pairwise differences between haplotypes, from which parameters of a demographic (NEW in ver 3.x) or spatial population expansion can be estimated |
|
Haplotype frequency estimation |
Estimates the frequency of haplotypes present in the population by maximum likelihood methods. |
|
Gametic phase estimation |
Estimates the most like gametic phase of multi-locus genotypes using a pseudo-Bayesian approach (ELB algorithm). |
|
Linkage disequilibrium |
Test of non-random association of alleles at different loci. |
|
Hardy-Weinberg equilibrium |
Test of non-random association of alleles within diploid individuals. |
|
Tajima’s neutrality test |
Test of the selective neutrality of a random sample of DNA sequences or RFLP haplotypes under the infinite site model. |
|
Fu's FS neutrality test |
Test of the selective neutrality of a random sample of DNA sequences or RFLP haplotypes under the infinite site model. |
|
Ewens-Watterson neutrality test |
Tests of selective neutrality based on Ewens sampling theory under the infinite alleles model. |
|
Chakraborty’s amalgamation test |
A test of selective neutrality and population homogeneity. This test can be used when sample heterogeneity is suspected. |
|
Minimum Spanning Network (MSN) |
Computes a Minimum Spanning Tree (MST) and Network (MSN) among haplotypes. This tree can also be computed for all the haplotypes found in different populations if activated under the AMOVA section. |
|
Inter-population methods: |
Short description: |
|
Search for shared haplotypes between populations |
Comparison of population samples for their haplotypic content. All the results are then summarized in a table. |
|
AMOVA |
Different hierarchical Analyses of Molecular Variance to evaluate the amount of population genetic structure. |
|
Pairwise genetic distances |
FST based genetic distances for short divergence time. |
|
Exact test of population differentiation |
Test of non-random distribution of haplotypes into population samples under the hypothesis of panmixia. |
|
Assignment test of genotypes |
Assignment of individual genotypes to particular populations according to estimated allele frequencies. |
|
Mantel test: |
Short description: |
|
Correlations or partial correlations between a set of 2 or 3 matrices |
Can be used to test for the presence of isolation-by-distance |
Windows 95/98/NT/2000/XP.
A minimum of 128 MB RAM, and more to avoid swapping.
At least 10Mb free hard disk space.
Download Arlequin31.zip to any temporary directory.
Extract all files contained in Arlequin31.zip in the directory of your choice.
Start Arlequin by double-clicking on the file WinArl3.exe, which is the main executable file.
Configure Arlequin: Choose which Text Editor to use when editing project files in the "Arlequin Configuration" tab.
The first thing to do before running Arlequin for the first time is certainly to read the manual. it will provide you with most of the information you are looking for. So, take some time to read it before you seriously start analyzing your data.
Version 3.0: Compared to version 2, Arlequin version 3 now integrates the core computational routines and the interface in a single program written in C++. Therefore Arlequin does not rely on Java anymore. This has two consequences: the new graphical interface is nicer and faster, but it is less portable than before. At the moment we release a Windows version (2000, XP, and above) and we shall probably release later a Linux. Support for the Mac has been discontinued.
Other main changes include:
Correction of many small bugs
Incorporation of two new methods to estimate gametic phase and haplotype frequencies
EM zipper algorithm: An extension of the EM algorithm allowing one to handle a larger number of polymorphic sites than the plain EM algorithm.
ELB algorithm: a pseudo-Bayesian approach to specifically estimate gametic phase in recombining sequences.
Incorporation of a least-square approach to estimate the parameters of an instantaneous spatial expansion from DNA sequence diversity within samples, and computations of bootstrap confidence intervals using coalescent simulations.
Estimation of confidence intervals for F-statistics, using a bootstrap approach when genetic data on more than 8 loci are available.
Update of the java-script routines in the output html files, making them fully compatible with Firefox 1.X.
A completely rewritten and more robust input file parsing procedure, giving more precise information on the location of potential syntax and format mistakes.
No need to define a web browser for consulting the results. Arlequin will automatically present the results in your default web browser (we recommend the use of Firefox freely available on http://www.mozilla.org/products/firefox/central.html.
Version 3.01: Compared to version 3.0, Arlequin 3.01 include some bug corrections and some additional features:
Bug corrections:
Minimum Spanning Tree Checkbox was not available for Genotypic data with known Gametic Phase.
Choice of how FST is computed was not available when computing pairwise distances. Now, it is synchronized with the choice of distance in the AMOVA panel.
"Search for shared haplotypes" did not work for Genotypic Data with known Gametic Phase. This has been corrected and Arlequin now ouputs a list of haplotypes before the table of frequencies.
[[Mantel]] section was not recognized if located after a [[Structure]] section.
Improved conversion between GenePop and Arlequin formats.
"Diploid Data" option is now present when converting from Genepop to Arlequin.
Output of s.d. of the number of alleles (k) was sometimes zero in output of Fu's FS test. This is now corrected and annoying warning messages about " "No molecular diversity within a sample while performing Fu's test" have been suppressed in output file.
Additions:
Version 3.1: Compared to version 3.01, Arlequin 3.1 includes cosmetic and speed improvements, several bug corrections and additional features:
Bug corrections:
Improvements
Additions:
Version 3.11: Compared to version 3.1, Arlequin 3.11 is mainly an update of ver 3.1, and there is no new manual.
Bug corrections:
Modifications
Additions:
Excoffier, L. G. Laval, and S. Schneider (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1:47-50.
Problems can be reported on the Arlequin Discussion Forum located on the Genetic Software Forum (GSF: http://www.rannala.org/gsf) on and hosted by Bruce Rannala.
This Arlequin forum will also be used as a Frequently Asked Questions (FAQ) page
Download Arlequin ver 3.11 for Windows (posted on 19.02.2007)
Download Arlequin 3.1 User Manual
 Configuration
|
 Project wizard
|
 Import-Export
|
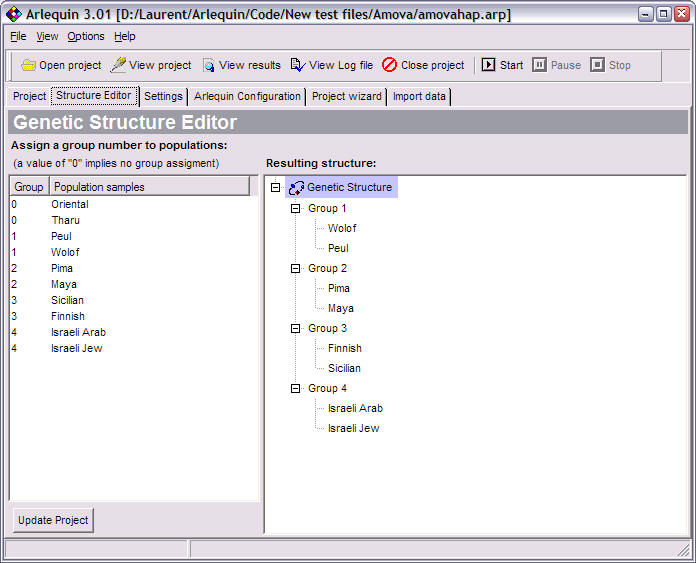 Genetic structure Editor |
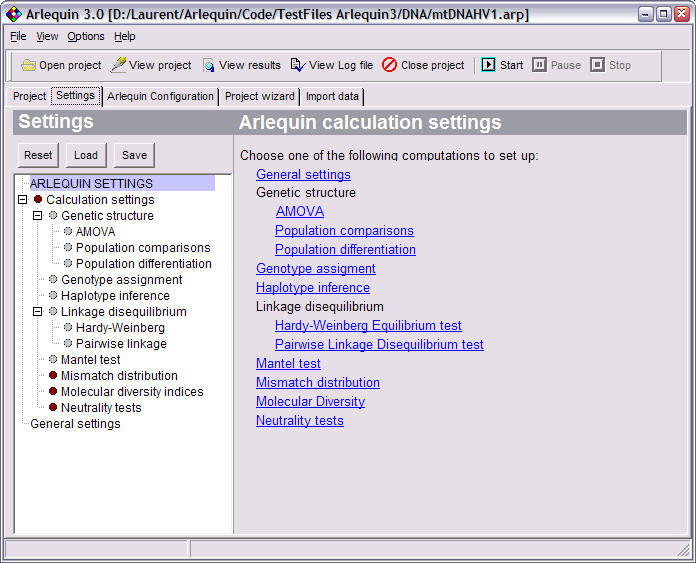 Arlequin settings
|
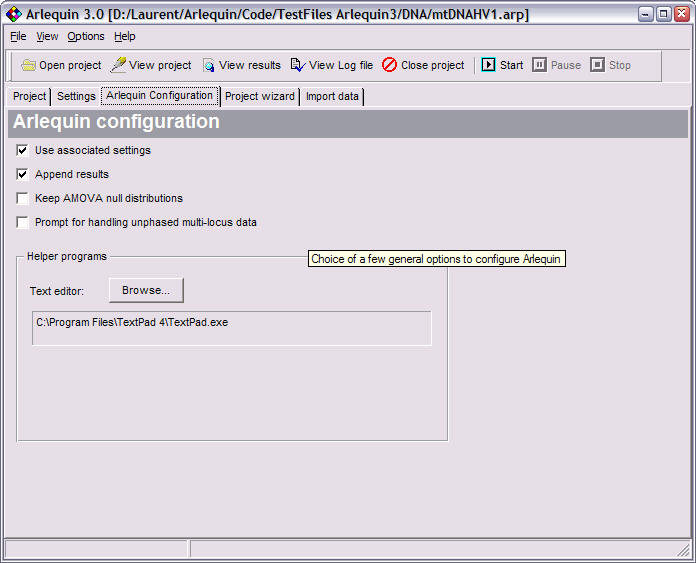 Arlequin configuration
|
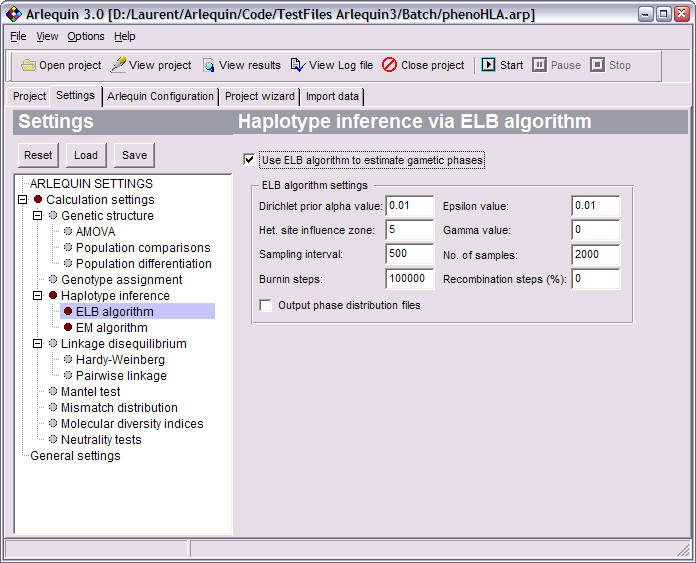 ELB settings
|
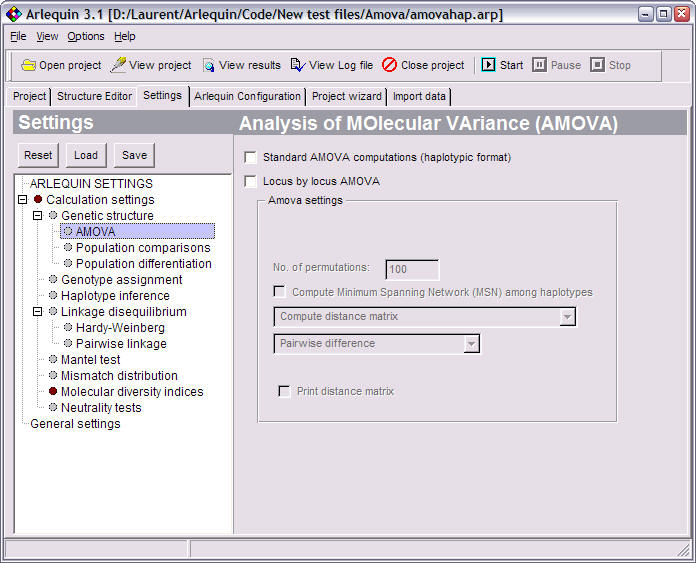 AMOVA settings
|
 LD settings
|
 Neutrality tests settings |
 Tabbed Output File in Firefox
|
File conversion programs
| Name |
Input format |
Output format |
|---|---|---|
| Convert | Excel | WhichRun, GeneClass, GDA, Microsat, Arlequin, Cervus, Fstat, Structure, Phylip |
| Formatomatic | Raw (csv), GenePop | Raw (csv), GenePop, Arlequin, Immanc |
| GenePop on the web | GenePop | Arlequin, Biosys, Fstat, Structure, Phylip |
Other free population
genetics software programs
| Program | Short description |
|---|---|
| Batwing | Bayesian Analysis of Trees With Internal Node Generation |
| BayesAss | Bayesian estimation of recent migration rates using multilocus genotypes |
| DnaSP | A software package for the analysis of nucleotide polymorphism from aligned DNA sequence data |
| Fstat |
Computer package which estimates and tests gene diversities and differentiation statistics from codominant genetic markers |
| GDA |
Computes linkage and Hardy-Weinberg disequilibrium, some genetic distances, and provides method-of-moments estimators for hierarchical F-statistics |
| GeneClass |
GeneClass is a program for assignation and exclusion using molecular markers |
|
GenePop,
GenePop on the web |
Software package for the computations of various population genetics parameters |
| Genetix |
Software package for the computations of various population genetics parameters |
| Hickory |
Bayesian estimation of F-statistics from dominant marker data |
| Immanc |
Detection of immigrant by using multilocus genotypes |
|
|
|
| Lamarc |
Suite of programs which estimate population-genetic parameters, based on McMC likelihood methods |
| Mark Beaumont's programs |
|
| Mega | Integrated tool for automatic and manual sequence alignment, inferring phylogenetic trees, mining web-based databases, estimating rates of molecular evolution, and testing evolutionary hypotheses |
| PAML |
A package of programs for phylogenetic analyses of DNA or protein sequences using maximum likelihood |
| Phase |
Program for haplotype reconstruction, and recombination rate estimation from population data |
| PopGene |
Analysis of genetic variation among and within populations using co-dominant and dominant markers |
| Populations |
Computation of various genetic distances |
| Structure |
Software package for using multi-locus genotype data to investigate population structure |
| WhichRun |
A computer program for population assignment of individuals based on multilocus genotype data |
A more extensive list of population genetics programs can be found here, here, here, and here.
We have also written a review on available programs in population genetics, which contains the description of about 25 different programs, with links to many more. It can be found here.
Laurent Excoffier, CMPG, Institute of Ecology and Evolution, University of Bern
Last edited on 17.02.07 (18:21)