
New version!Download SPLATCHE 1.1(for Windows XP, 2000) |
SPLATCHE (for SPatiaL And Temporal Coalescences in Heterogenous Environment) is a program that allows to incorporate the influence of environment in the simulation of migration of a given species from one origin. In a second phase, the molecular genetic diversity of one or several samples drawn from the simulated species can be generated.
Geographic area and environmental information have to be specified by the program user in a series of input files. Basically, the virtual world where migrations take place is constituted by a matrix of demes. Each deme has its own environmental characteristics according to the input files. A coalescent-based approach allows to generate the molecular diversity of any population sample. The molecular data obtained can then be analyzed in order to study the signature of the simulated demographic scenario.
The goal of this online manual is to describe the technical aspects of the
software SPLATCHE (version 1.1). This manual complements the article from Currat,
Ray and Excoffier, published in 2004. Further details on the methodology can
also be found in Ray (2003) and Currat (2004). The pdf version of the user manual
could also be download there.
In version 1.1, some input files have changed and are not backward compatible with SPLATCHE 1.0. Please see the User Manual for details.
The old webpage of version 1.0 can still be accessed here.
This software was developped by Mathias Currat, Nicolas Ray and Laurent Excoffier and the reference to cite is:
Currat M, Ray N, & Excoffier L (2004) SPLATCHE: a program to simulate genetic diversity taking into account environmental heterogeneity, Molecular Ecology Notes 4(1): 139-142 [PDF file]
| User manual |
| Download SPLATCHE v. 1.1 user manual (PDF) |
Publications using SPLATCHE Below is a list of selected papers having used SPLATCHE. For a complete list of papers referencing the SPLATCHE paper, click on this Google Scholar link. |
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Molecular Biology and Evolution 20(1):76-86 [PDF file]
Currat M, Excoffier L (2004) Modern Humans Did Not Admix with Neanderthals during Their Range Expansion into Europe. PLoS Biol 2(12): e421 [PDF file]
Currat M, Excoffier L (2005) The effect of the Neolithic expansion on European molecular diversity. Proc. R. Soc. Biological Sciences 272: 679-688
Hamilton G, Currat M, Ray N, Heckel G, Beaumont M, Excoffier L (2005) Bayesian estimation of recent migration rates after a spatial expansion. Genetics 170: 409-417 [Link]
Hamilton G, Stoneking M, Excoffier L (2005) Molecular analysis reveals tighter social regulation of immigration in patrilocal populations than in matrilocal populations. Proc Natl Acad Sci USA 102(21): 7476-7480
Ray N, Currat M, Berthier P, Excoffier L (2005) Recovering the geographic origin of early modern humans by realistic and spatially explicit simulations. Genome Research 15: 1161-1167 [Link]
Klopfstein S, Currat M, Excoffier L (2006) The fate of mutations surfing on the wave of a range expansion. Mol. Biol. Evol. 23: 482-490 [Link]
Foll M, Gaggiotti, O (2006) Identifying the Environmental Factors That Determine the Genetic Structure of Populations. Genetics 174: 875-891 [Link]
Currat M, Excoffier L, Maddison W, Otto SP, Ray N, Whitlock MC, Yeaman S (2006) Comment on "Ongoing Adaptive Evolution of ASPM, a Brain Size Determinant in Homo sapiens" and "Microcephalin, a Gene Regulating Brain Size, Continues to Evolve Adaptively in Humans. Science 313(5784): 172 [Link]
Wegmann D, Currat M, Excoffier L (2006) Molecular Diversity After A Range Expansion In Heterogeneous Environments. Genetics in press [Link]
Excoffier L & Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends in Ecology and Evolution 23(7): 347-351 [Abstract and pdf]
Currat M, Ruedi M, Petit RJ & Excoffier L (2008) The hidden side of invasions: massive introgression by local genes. Evolution, in press [Abstract and pdf]
Principles:
The demographic and spatial expansion module allows to simulate a demographic
and spatial expansion from one or many initial populations. The simulation uses
discrete time and space. The unit of time is the generation, while the unit
of the 2D space is a cell, also called a deme. Each deme has the same size and
can be considered as a homogeneous subpopulation. The spatial model used in
SPLATCHE is the 2D stepping-stone model (Kimura and Weiss 1964), which defines
a regularly spaced array of demes. Each deme undergoes an independent population
growth and can exchange emigrants with its four direct neighboring demes.
Each deme is also considered as a sub-unit of the environment. The environment
can influence the local demography through its carrying capacity (maximum number
of individuals) and its friction (facility to migrate through). These two environmental
characteristics can be defined for the entire array of demes through input maps.
Variations through time of carrying capacity and/or friction values are also
possible, which is defined as a dynamic environment.
Available demographic models:
The logistic population growth of each deme follows a standard logistic curve,
of the form
![]()
where K is the carrying capacity, and r is the growth rate.
For the migration part of the demography, three models are available in SPLATCHE:
-Model 1. Migration model with even number of emigrants
The number of emigrants M from a deme is computed, for each generation, as M=mNt,
where m is the migration rate, and Nt is the population density of the deme
at generation t. The number of emigrants Mt in any of the four directions is
then computed as
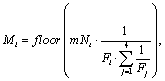
-Model 2. Migration model with absolute number of emigrants
Same as Model 1, but the fractional part of Mt is not truncated. Instead, a
multinomial distribution is used to split M emigrants to the neighboring demes
(see Ray 2003). This ensures that there are always M emigrants that are sent.
The drawback of this technique is that it requires the drawing of random numbers,
which increases the time required for a simulation.
-Model 3. Stochastic migration model with absolute number of emigrants
Same as Model 2, but the number of emigrants M varies stochastically as a Poisson
variable centered around mNt.
General Settings panel
The General Settings panel is the primary panel to set the demographic parameters
and to launch a demographic simulation. A screenshot of this panel is shown
in Figure 1. A description of each component of this panel is given below.
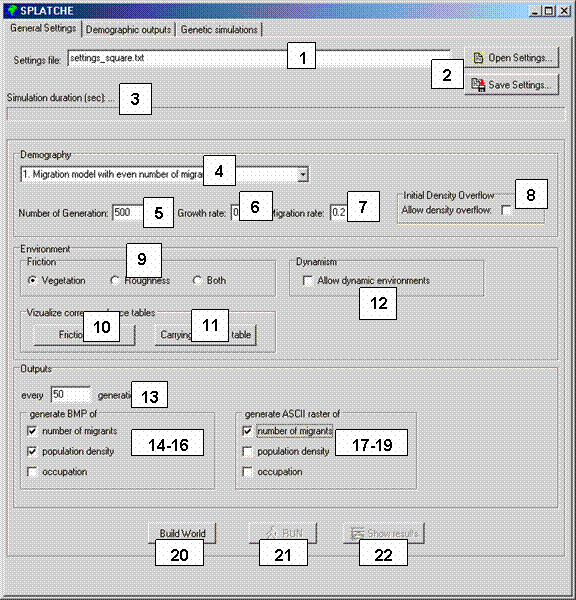
Figure 1. General Setting panel. The numbers correspond to a description in the text
- General
[ 1 ] Settings file name: location of the settings file (*.txt). See here
for the full description of a settings file.
[ 2 ] Buttons allowing to open a settings file or to save a settings file.
[ 3 ] Progress bar showing the remaining computation time of a current simulation.
The duration of a simulation (in seconds) is also given at the end of the computation.
- Demography related parameters
[ 4 ] Drop-down menu allowing to choose among the three available demographic
models.
[ 5 ] Number of simulated generations. The generation time is the number of
time units par generation. It can be set in order to get the "real time"
while browsing the results in the "Graphical outputs" window.
[ 6 ] Growth rate used in the demographic models. This is the net growth rate
used in the logistic growth phase.
[ 7 ] Migration rate used in the demographic models. The migration rate is the
fraction of the deme population that will migrate out at each generation. For
a deme population of size , the number of emigrants is then equal to at each
generation.
[ 8 ] Checkbox to allow the initial density overflow. If this checkbox is switched
on and the size of the initial population exceeds the carrying capacity of the
deme, the initial population is spread over neighboring demes until all the
individuals are placed in a deme. The overflow function fills a deme at carrying
capacity before using neighboring demes. If this checkbox is switched off, the
size of the initial population is always the size sets in the initial density
file. See here for more details.
- Environment related parameters
[ 9 ] Radio button allowing to choose how the friction values are computed.
When "vegetation" or "roughness" is chosen, friction values
are only computed from the corresponding input data set (see here).
If "both" is chosen, friction values are computed by taking, for each
deme, the mean value between the friction value from the vegetation data set
and the friction value from the roughness data set.
[ 10 ] Button allowing to open the friction corresponding table (see here
for a description of this table) in the default text editor. The file can then
be modified and saved. The world must be rebuilt after a change in this file.
[ 11 ] Button allowing to open the carrying capacity corresponding table (see
here for a description of this table) in the default text
editor. The file can then be modified and saved. The world must be rebuilt after
a change in this file.
[ 12 ] CheckBox allowing a dynamic simulation (see here).
The world must be rebuilt after a change in this checkbox.
- Output parameters
Some output parameters are placed in this panel, because they need to be set
prior to a simulation, if one wants to automatically generate these outputs
during the simulation. These outputs are a temporal series of graphical representations
of the state of a demographic parameter (number of emigrants, population densities,
or occupation). Windows Bitmaps (BMP) or ASCII raster can be generated. The
output files are placed in two folders (called respectively, "BMP"
and "ASCII") which are created in the same folder than the setting
file. The filename of each output file is composed by the name of the demographic
variable followed by the number of generation at which it has been created.
[ 13 ] Number of generations between each output files. Beside the outputs for
the intermediate states, a series has always outputs for the initial and the
final state of the simulation.
[ 14-16 ] Checkboxes for the generation of BMP files.
[ 17-19 ] Checkboxes for the generation of ASCII raster files.
- Main buttons
[ 20 ] Button to build a world. It is during a building process that memory
space is allocated, and that carrying capacity and friction values are computed
for each deme
[ 21 ] Button to launch a simulation. If this button is grayed out, it means
that the world needs to be built or rebuilt.
[ 22 ] Button to show the graphical output window.
Graphical output window
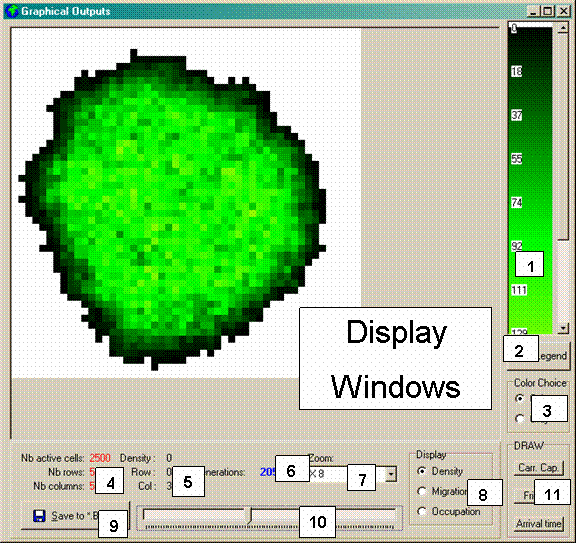
Figure 2. Graphical Outputs panel. The numbers correspond to
a description in the text.
[ 1 ] Legend for the current display.
[ 2 ] Buttons allowing to save the legend as a bitmap.
[ 3 ] Radio button for the choice of color or shades of gray display.
[ 4 ] Information on the number of active cells (cells having information for
the vegetation), the number of rows and the number of columns.
[ 5 ] Information on the density, the number of rows and the number of columns
when the mouse cursor is over a particular deme.
[ 6 ] Number of generations for the current display.
[ 7 ] Zoom for the current display.
[ 8 ] Radio button to choose from displaying the density, the number of emigrants,
or the occupation (black if occupied).
[ 9 ] Button allowing to save the current display as a bitmap.
[ 10 ] Cursor allowing to change the current generation, and the display at
the chosen generation.
[ 11 ] Buttons allowing to display the initial (at generation 0) carrying capacity
map, the initial friction map, and the proportional arrival time in each deme.
Demographic outputs window
This window allows to explore the demographic database that has been generated
through a simulation.
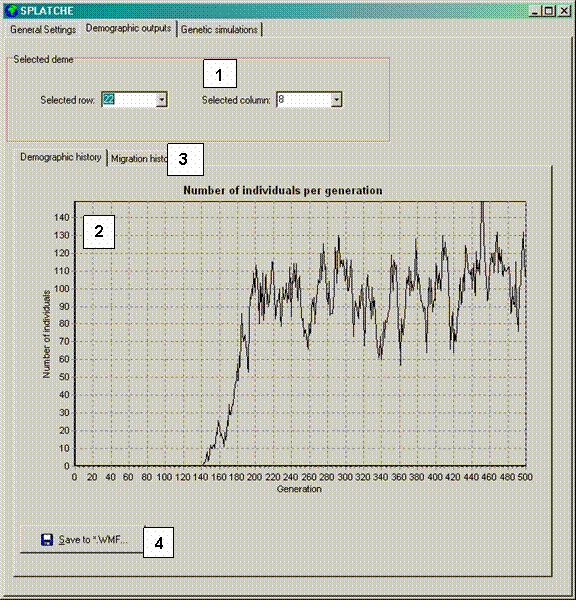
Figure 3. Demographic outputs panel. The numbers correspond to a description in the text.
[ 1 ] Selectors allowing to change the row and the column, which will select
the correct deme and display the history of the number of individuals (density).
[ 2 ] Graph showing the history of the number of individuals (density) for the
selected deme. It is possible to zoom in and out in the graph by drawing rectangles
with the mouse cursor (left button down).
[ 3 ] Second panel showing the histories of the number of emigrants in the four
directions.
[ 4 ] Button allowing to save the graph in Windows Metafile format.
Principles
Genetic simulations are always preceded by a demographic simulation. Indeed,
a genetic simulation uses the demographic information stored in the data base
generated during the demographic phase. The genetic phase is based on the "coalescent
theory", initially described by Kingman (Kingman 1982; Kingman 1982) and
developed in other papers (Ewens 1990; Hudson 1990; Donnelly and Tavaré
1995). This theory allows the reconstruction of the genealogy of a series of
sampled genes until their most recent common ancestor (MRCA). For neutral genes,
the genealogy essentially depends on the demographic factors that have influenced
the history of the populations from whom the genes are drawn. The implementation
of the coalescent theory is a modified version of SIMCOAL (Excoffier, Novembre
et al. 2000). The principal difference with SIMCOAL is that the demographic
information used by genetic simulations do not come anymore from the "migration
matrix" and "historical events", but from the data base generated
during the demographic simulation.
The genetic simulation itself follows the procedure described in (Excoffier et al. 2000) and consists in two phases:
1°) Reconstruction of the genealogy:
The reconstruction of the genealogy is independent of the mutational process.
Basically, a number n of genes is chosen. These genes are only identified by
their number and they have no genetic variability during this first phase. All
the n genes are associated with a geographic position in the virtual world where
the demography is simulated. These genes could belong to different demes in
the world. Then, going backward in time, the genealogy of these genes is reconstructed
until their most recent common ancestor (MRCA) in the following way:
Going backward in time, at each generation, two events can occur:
- Coalescent event: if at least two genes are on the same deme, they have a
probability to have a common ancestor at the preceding generation (a coalescent
event). This probability depends on the population size of the deme where the
genes are located. Each pair of genes has a probability 1/ Ni of coalescence
(if Ni is the number of haploid individual in the deme i). If there are ni genes
on the deme then the probability of one coalescent event becomes ni (ni -1)/
2Ni. Only one coalescent event is allowed per deme and per generation (see Ray,
Currat et al. 2003) for a discussion about this assumption).
- Migration: Each gene could have arrived with an immigrant from a different
deme. When going back in time, it means that the gene could leave the current
deme with the immigrant. So, the probability of migration from a deme i to a
deme j for a gene depends on the number of individuals that have arrived from
deme j to deme i at this generation. For each gene belonging to the deme i,
the probability of migration from deme j is equal to mji/Ni where mji is the
number of immigrants from deme j to deme i during the demographic phase.
All the deme sizes and the numbers of immigrant between demes are taken from
the database generated during the demographic simulation.
2°) Generation of the genetic diversity:
The second phase of a genetic simulation consists in generating the genetic
diversity of the samples. This operation is done in adding mutations independently
on all branches of the genealogy assuming a uniform and constant Poisson process.
At the end of this process all the sampled genes have a specific genetic identity.
The genetic process is entirely stochastic, so many genetic simulations have
to be performed for each demographic simulation in order to obtain meaningful
statistics. We recommend at least 1'000 simulations per demographic scenario.
The coalescent backward approach does not generate the history of the whole
population, but only that of sampled genes and their ancestors. Thus this approach
is much less demanding in terms of memory and computing time. That allows the
simulation of complex demographic scenarios within a very broad geographical
and temporal framework.
Genetic settings panel
Various parameters must be defined before launching a genetic simulation. The
number of parameters can be seen in Figure 4
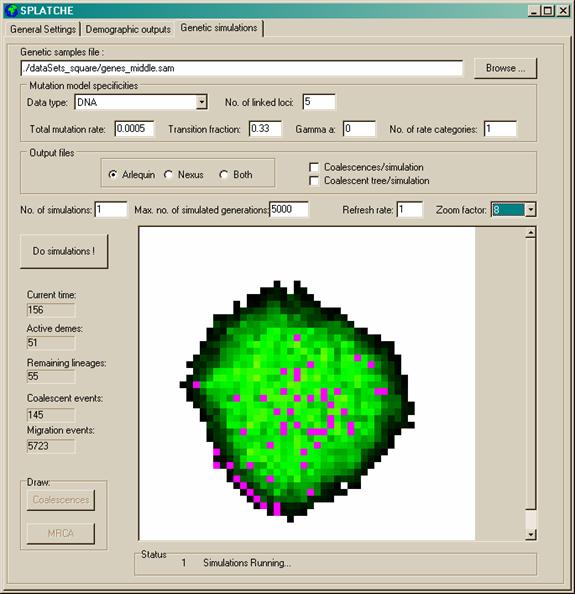
Figure 4 Genetic module panel. Demes where at least one gene
is present appear in violet.
- General
[ 1 ] Sample file name: location of the *.sam file.
[ 2 ] Number of simulations to be carried out
[ 3 ] Maximum of generations after which the process stop if the genealogy has
not been correctly reconstructed.
[ 4 ] Refresh rate: generation numbers after which the display window is updated.
[ 5 ] Zoom factor of the display window.
- Mutation model specificities
For all kind of data:
[ 6 ] Type of genetic data to be generated. It could be DNA, RFLP, Microsatellite
or Standard. See here for more details.
[ 7 ] Number of fully linked loci to simulate. It corresponds to the sequence
length for DNA.
[ 8 ] Mutation rate per generation for all loci taken together.
Specific to DNA:
[ 9 ] Transition bias: percentage of substitutions that are transitions.
[ 10 ] Gamma a: amount of heterogeneity in mutation rates along the sequence
according to either a discrete or continuous gamma distribution.
[ 11 ] Number of categories for DNA mutation variation.
Specific to Microsatellite:
[ 12 ]Range constraint: minimum and maximum size for microsatellite.
Genetic data
Different types of molecular data could be generated (RFLP, DNA, Microsatellites
and Standard), each with its own specificities:
-RFLP data: Only a pure 2-allele model is implemented. Several fully linked RFLP loci can be simulated, assuming a homogeneous mutational process over all loci. A finite-sites model is used, and mutations can hit the same site several times, switching the RFLP site on and off. We thus assume that there is the same probability for a site loss or for a site gain.
-Microsatellite data: We have implemented a pure stepwise mutation model
(SMM) with or without constraint on the total size of the microsatellite. Several
fully linked microsatellite loci can be simulated under the same mutation model
constraints. The output for each loci is listed as a number of repeat, having
started arbitrarily at 10,000 repeats. The number of repeats for each gene should
thus be centered around that value.
-DNA sequence data: We have implemented here several simple finite-sites
mutational models. The user can specify the percentage of substitutions that
are transitions (the transition bias), the amount of heterogeneity in mutation
rates along a DNA sequence according to either a discrete or continuous Gamma
distribution. We can therefore simulate DNA sequences under a Jukes and Cantor
model (Jukes and Cantor 1969) or under a Kimura-2-parameter model (Kimura 1980),
with or without Gamma correction for heterogeneity of mutation rates (Jin and
Nei 1990). Other mutation models that depend on the nucleotide composition of
the sequence were not considered here, because of their complexity and because
they require specifying many additional parameters, like the mutation transition
matrix and the equilibrium nucleotide composition.
-Standard data: Following the definition given in Arlequin User Manual
(Schneider et al. 2000), this type defines data for which the molecular basis
is not particularly defined, such as mere allele frequencies. The comparison
between alleles is done at each locus. For each locus, the alleles could be
either similar or different.
Initial density and origin location
!! VERSION 1.1: this file format has changed and is not backward compatible with SPLATCHE 1.0
A file, called “dens_init.txt” in the examples, is used to specify the place(s)
of origin of the simulated population. This file contains a first line indicating
the number of origins, followed by one line per origin, and finally by a legend.
Each line per origin contains 6 fields separated by “tab” or “space” character:
1. Name of the source population (do not use space characters for this name!).
2. Size of the source population, in number of effective haploid individuals.
3. & 4. Geographic coordinates of the population source (latitude and longitude).
SPLATCHE will determine itself in which particular deme corresponds the coordinates
of the population. Coordinates must belong to the geographical surface defined
in the header of the environmental files. Coordinates do not need to be in a
particular units (e.g. decimal degrees), but they needs to be in the same units
that the coordinates defined in the header of the environmental files.
5. Resize parameter: it is the size of the population source before the beginning
of the expansion. This parameter is used only for genetic simulations. If this
parameter is set to 0, then the size of the population source before the onset
of the expansion is regarded as being equal to the initial size (parameter 2.).
In case "initial density overflow" is switch on, the Resize parameter
must be set to the total size of the initial population (e.g. 2'000) if the
user wants to keep this initial size before the beginning of the expansion.
6. Migration rate from origin: this parameter is only used with genetic simulations
when there more than one origin. It is the emigration rate from the current
origin to the other origin(s) during the time prior to the expansion. It is
also equivalent to the probability of a lineage emigrating from the current
origin. When a lineage emigrate toward more than one origin, it is dispatched
randomly to one of the target origins.
Two examples of initial density files, with 1 or 2 origins:
The Figure below shows a scheme of the simulation process with one or three origins. The parameter "Tau" (set trough the settings file) indicates the duration (in years) during which the population is kept at a size equal to the "Resize" parameter. After that time, all remaining lineages are put in a single deme of size 100 until the ultimate coalescent event.
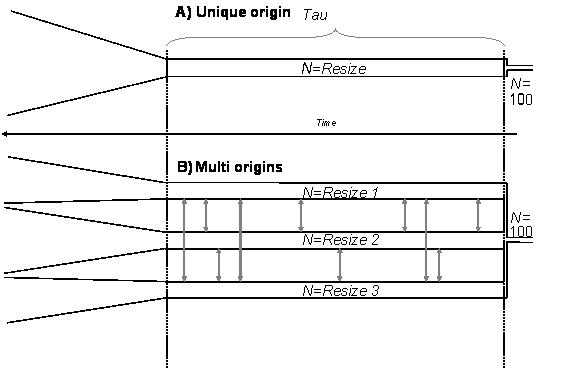
Scheme of the unique or multiple origins models
Settings file
!! VERSION 1.1: this setting file has changed and is not backward compatible
with SPLATCHE 1.0
All parameters (unless "generation time" and "tau") can
be defined using the graphical interface of SPLATCHE. However, it is possible
to save parameters into a new settings file, so that they can be recovered later.
Only the graphical parameters are not contained in the settings files. An example
of settings files is provided with SPLATCHE: “settings_square.txt”, with the
corresponding data files in the folder called "dataSets_square". The
example file is a simple square world constituted by 50x50 demes (see Ray et
al. 2003).
The setting file is composed of 31 parameters. An example, corresponding to
"settings_square.txt", is given below. Each line starts with the value
of the parameter, followed by a blank, a double slash, and then the description
of the parameter.
Parameters in bold indicate new parameters compared to SPLATCHE v. 1.0
ASCII format for environmental data
The environmental datasets that can be loaded into SPLATCH must be in ASCII
raster format. Two different datasets can be loaded. The first one is the "vegetation"
dataset, defining to what type (category) of vegetation belongs each deme. The
second dataset is the "roughness" dataset, defining continuous friction
values, such as friction computed from topography.
This format of the environmental dataset is composed of a header (first six
lines) containing information on the file, then a matrix of values in rows and
columns.
The header information is as follow:
ncols : number of columns
nrows : number of rows
xllcorner : longitude coordinate of the lower-left deme
yllcorner : latitude coordinate of the lower-left deme
cellsize : width of a deme (cell size), in same units than the coordinates
NODATA_value : value indicating than a deme must not be considered (like sea)
Example of an environmental dataset

Dynamic simulations and conversion tables to obtain K and F
It is possible in SPLATCH to do dynamic simulations. A dynamic simulation allows variation of carrying capacity and/or friction value at different time during the course of a simulation. In order to set at what time the changes occur, different files are needed.
The two main files, which are set through the settings files, are typically called "Dynamic_K.txt" and "Dynamic_F.txt". On the first line of each of this file appears the number of changes during a simulation. Then each line (one per change) is composed by the time of change (in generations), the filename of the corresponding table (see below), and an arbitrary description. The three components of each line must be separated by a blank space. For a non-dynamic simulation, only the first filename is considered, regardless of the number indicated on the first line.
Example of "Dynamic_K.txt" file:

Each file name must target a valid "conversion table" that makes the link between a particular vegetation (or land cover) category and a carrying capacity (or friction) value. A conversion table is composed of a vegetation category number, followed by a carrying capacity (or friction) value, and by a description. The vegetation category numbers must correspond to the numbers found in the input "vegetation" dataset (see previous chapter).
Example of "veg2K.txt" file:

By having several corresponding tables for the carrying capacity and/or the friction values, it is then possible to simulate complex changes of the environment through time.
Genetic samples
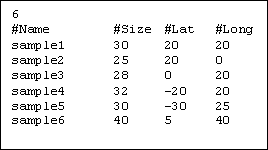
A file with the extansion ".sam" allows to specify the localization
of the population sampled, as well as the number of genes sampled in each population
(see Figure 4)
On the first line of this file, the user can specify the number (integer) of
population sampled. The second line is reserved for the legends. Then, each
line defines a sample with 4 fields separated by "tab" our "space"
character.
1. Name of the population from which the sample has been drawn.
2. Number of genes belonging to that sample.
3. & 4. Geographic location of the population. (latitude and longitude).
SPLATCHE will determine automatically in which particular deme falls the coordinates
of the population. The coordinates must belong to the geographical surface defined
in the header file.
Genetic output file choice [ 13 ] and [ 14 ] on Figure 4
Various kinds of genetic output files can be generated by SPLATCHE:
Arlequin files
The genetic data generated by one simulation are directly output in an ARLEQUIN
project file, with the extension “*.arp”. This file format allows one to compute
the data using the ARLEQUIN software in order to obtain different statistics,
see ARLEQUIN manual (Schneider et al. 2000) for more details. If more than one
simulation is performed using one demographic simulation (which is usually the
case) then an ARLEQUIN batch file (with extension “*.arb”) is also generated,
listing all simulated files, and allowing one to compute statistics on the whole
set of simulated files. Note also that the ARLEQUIN software has a file conversion
utility for exporting input data files into several other format like BIOSYS,
PHYLIP, or GENEPOP, so that files produced by SPLATCHE could be also analyzed
by these softwares after file conversion.
Nexus files
Two other types of file produced by Friction are compatible with the NEXUS file
format: for each simulation, a file with “*paup” extension could be generated.
This file lists all the simulated genes together with their true genealogical
structure. This file can be analyzed with David Swofford's PAUP* software (1999).
A PAUP batch file, with extension “*.bat” is also generated.
Coalescence distribution files
A bitmap representing the spatial distribution of the coalescent events for
all the simulations joined is automatically created with the “*_TotNumCoal.bmp”
termination. This bitmap can also be visualized through the button “Draw Coalescence”
(15 on Figure 4.1) on the interface. When checking the coalescence checkbox
(16 on Figure 4.1), similar bitmaps of the spatial distribution of coalescent
events are generated for every simulations (with the “*_NumCoal.bmp” termination).
The times for each coalescent event and each simulation are listed on a file
with “*.coal” extension. Those times are given in generation units, with larger
numbers corresponding to the end time of the simulation.
Coalescent trees files
By checking the checkbox “coalescent trees” (16 on Figure 4.1), it is possible
to generate for each simulation a bitmap representing the genealogical links
between each node of the coalescence tree, laid out spatially. Those files are
terminated with “*_CoalTree_*.bmp”.
MRCA files
SPLATCHE gives information on the localization and timing of the Most Recent
Common Ancestor (MRCA) of the totality of genes sampled, and the MRCA of each
of the various samples. A file with the termination “*_MRCADensity.bmp” is automatically
generated and is a bitmap of the spatial distribution of MRCA for all the simulations
added together. These maps can also be visualized through the button “Draw MRCA”
(15 on Figure 4.1) on the interface. Similar bitmaps, with the “*_MRCAPopDensity*.bmp”
termination, are generated for each sample. The Time to the Most Recent Common
Ancestor (TMRCA) for the whole tree and for each sample are also listed in a
file with the “*.tmrca” extension. The TMRCA are given in generation units,
with larger numbers corresponding to the end time of the simulation.
Tree files
Two files with the “*.trees” extension are automatically produced and list all
the simulated trees, with branch lengths expressed either i) in units of generations
scaled by the population size (N), and therefore representing the true coalescent
history of the sample of genes, or ii) in units of average number of substitutions
per site, and therefore representing the realized mutational tree. These two
files could be visualized with the software TREEVIEW (Page 1996).
We are grateful to Stefan Schneider and Pierre Berthier for their computing
assistance. The development of the SPLATCHE program was possible through Swiss
NSF grants n° 31-054059.98 and 3100A0-100800
-Currat, M. (2004). Effets des expansions des populations humaines en Europe
sur leur diversité génétique. Département d'Anthropologie
et Ecologie. Genève, Université de Genève.
-Currat, M., N. Ray, et al. (2004). SPLATCHE: a program to simulate genetic
diversity taking into account environmental heterogeneity, Molecular Ecology
Notes, Volume 4, Issue 1, Page 139-142
-Donnelly, P. and S. Tavaré (1995). "Coalescents and genealogical structure under neutrality." Annu. Rev. Genet. 29: 401-421.
-Ewens, W. J. (1990). Population Genetics Theory - The Past and the Future. Mathematical and Statistical developments of Evolutionary Theory. S. Lessar. Dordrecht, Kluwer Academic Publishers: 177-227.
-Excoffier, L., J. Novembre, et al. (2000). "SIMCOAL: A general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography." J. Heredity 91: 506-510.
-Hudson, R. (1990). Gene genealogies and the coalescent process. oxford, Oxford University Press.
-Jin, L. and M. Nei (1990). "Limitations of the evolutionary parsimony method of phylogenetic analysis." Mol. Biol. Evol. 7: 82-102.
-Jukes, T. and C. Cantor (1969). Evolution of protein molecules. Mamalian Protein Metabolism. H. N. Munro. New York, Academic press: 21-132.
-Kimura, M. (1980). "A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences." J. Mol. Evol. 16: 111-120.
-Kimura, M. and W. H. Weiss (1964). "The stepping stone model of genetic structure and the decrease of genetic correlation with distance." Genetics 49: 561-576.
-Kingman, J. F. C. (1982). "The coalescent." Stoch. Proc. Appl. 13: 235-248.
-Kingman, J. F. C. (1982). "On the genealogy of large populations." J. Appl. Proba. 19A: 27-43.
-Page, R. D. M. (1996). "TREEVIEW: An application to display phylogenetic trees on personal computers." Comput. Appl. Biosci. 12: 357-358.
-Ray, N. (2003). Modélisation de la démographie des populations humaines préhistoriques à l'aide de données environnementales et génétiques. Thèse, Départment d'Anthropologie. Genève, Université de Genève: 331.
-Ray, N., M. Currat, et al. (2003). "Intra-deme molecular diversity in spatially expanding populations." Molecular Biology and Evolution 20(1): 76-86.
-Schneider, S., D. Roessli, et al. (2000). Arlequin: a software for population genetics data analysis. User manual ver 2.000. Geneva, Genetics and Biometry Lab, Dept. of Anthropology, University of Geneva.
Contact: Nicolas Ray, Computational and Molecular Population Genetics Lab, University of Bern
Last edited on 15.05.05