 |
 |
fast sequential Markov coalescent simulation of genomic data under complex evolutionary models |
benchmarks
We have performed a few comparisons of fastsimcoal with ms, simcoal2 or MaCSspeed
datasets
The following test data sets were used in our speed comparisons|
|
No. of populations |
Diploid
population size |
Migration rate |
Mutation rate |
Recombination rate |
Sample size per
population |
|
1popNoRec |
1 |
12500 |
|
2×10-8 |
0 |
2000 |
|
1popSmallSample |
1 |
12500 |
|
2×10-8 |
1.2×10-8 |
20 |
|
1popLargeSample |
1 |
12500 |
|
2×10-8 |
1.2×10-8 |
2000 |
|
2popNoRec |
2 |
6250 |
0.001 |
2×10-8 |
0 |
1000 |
|
2popSmallSample |
2 |
6250 |
0.001 |
2×10-8 |
1.2×10-8 |
10 |
|
2popLargeSample |
2 |
6250 |
0.001 |
2×10-8 |
1.2×10-8 |
1000 |
The population, mutation and recombination parameters correspond to those used by Chen et al. (2009) in their comparison of ms to MaCS.
results
The results reported below are average CPU time per run expressed in
seconds. All runs were run on a Linux cluster made up of 2.6GHz AMD
Opterons with 4 GB of RAM and 74 GB HD.|
Data set |
No. of replicates |
Sequence length |
Program |
||
|
ms |
MaCS |
fastsimcoal |
|||
|
1popNoRec |
1000 |
1 Mb |
1.1 |
11.1 |
9.5 |
|
100 |
10 Mb |
9.6 |
107.0 |
72.9 |
|
|
100 |
100 Mb |
147.9 |
1319.5 |
1038.1 |
|
|
2popNoRec |
1000 |
1 Mb |
1.2 |
12.5 |
9.3 |
|
100 |
10 Mb |
8.9 |
128.1 |
71.5 |
|
|
100 |
100 Mb |
161.2 |
1513.2 |
1099.9 |
|
Without recombination, ms is much faster than the two other programs based on the SMC’ approximation, and fastsimcoal is becomes increasingly faster than MaCS with larger recombination rates and with migration.
|
Data set |
No. of replicates |
Sequence length |
Program |
||
|
ms |
MaCS |
fastsimcoal |
|||
|
1popSmallSample |
1000 |
1 Mb |
0.344 |
0.242 |
0.095 |
|
100 |
10 Mb |
159.246 |
2.618 |
0.460 |
|
|
100 |
100 Mb |
x |
26.124 |
4.364 |
|
|
2popSmallSample |
1000 |
1 Mb |
0.378 |
0.907 |
0.152 |
|
100 |
10 Mb |
165.507 |
9.094 |
1.080 |
|
|
100 |
100 Mb |
x |
97.876 |
10.559 |
|
x : ms crashed
For small sample sizes (total n=20) and with recombination, the SMC’
based programs are becoming much faster than ms, which
fails to run for 100Mb sequences. For such small sample
sizes, fastsimcoal is 2.5 to 9.3
times faster than MaCS. For MaCS and fastsimcoal, computing time increases approximately
linearly with
sequence length, as expected.
|
Data set |
No. of replicates |
Sequence length |
Program |
||
|
ms |
MaCS |
fastsimcoal |
|||
|
1popLargeSample |
1000 |
1 Mb |
3.7 |
28.1 |
25.2 |
|
100 |
10 Mb |
x |
327.5 |
235.7 |
|
|
100 |
100 Mb |
x |
3700.8 |
2635.4 |
|
|
2popLargeSample |
1000 |
1 Mb |
3.9 |
33.3 |
25.9 |
|
100 |
10 Mb |
x |
393.5 |
240.6 |
|
|
100 |
100 Mb |
x |
4311.1 |
2684.7 |
|
x : ms crashed
For large sample sizes (total n=2000), ms is actually faster than the two other programs for a “small” sequence of 1Mb, but failed to run successfully for longer sequences. For these large sample sizes, fastsimcoal is 1.2 to 1.8 times faster than MaCs. fastsimcoal computing time still increases approximately linearly with sequence length, which is not the case ofMaCS, which becomes slightly penalized by larger sequences. Note however, that for 1Mb and 10Mb, we used fastsimcoal options allowing it to keep all simulated sites in memory before writing them to the output file, which was not possible for 100Mb sequences, which would use up too much memory.
patterns of molecular diversity
Number of pairwise difference
We report below a comparison of the patterns of diversity within and between populations simulated by ms, MaCs and fastsimcoal.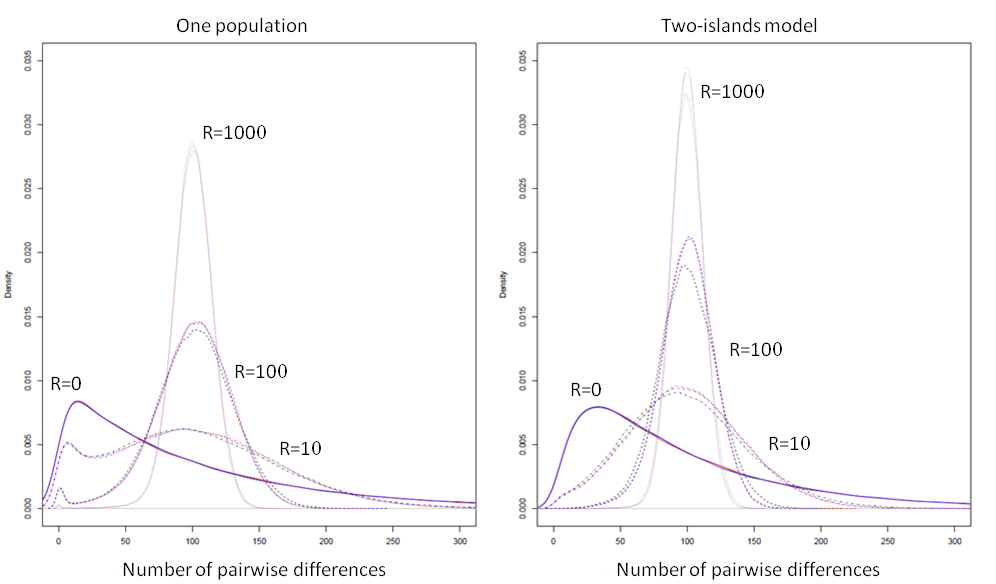
ms results are shown with a black line, MaCs with a red line, and fastsimoal, with a blue line. In all cases, the empirical distributions of the number of pairwise differences were computed from 100,000 simulations of the coalescent of 2 genes. The 2 genes were drawn from a single population for the one-population case, and were drawn each in a different population in the two-island model case. We used the following population parameters: for the one population case; and for the two-island case; and was varied between 0 and 1000, as shown above. In all cases, MaCS and fastsimcoal lead to identical distributions, which is expected as they are both based on the same SMC’ approximation.
In absence of recombination MaCS and fastsimcoal give also exactly the same distributions as ms, but are just running 7-10 times slower, as was seen in the previous section. With very high recombination rates, the SMC-based approximation of MaCS and fastsimcoal is extremely close to the ancestral recombination graph (ARG) implemented in ms, in keeping with previous results (McVean and Cardin, 2005). For "intermediate" recombination rates (R=10, R=100), some slight differences do emerge between ARG- and SMC-based programs, and these differences are slightly more pronounced in the 2-island model. However, it seems that these differences are much less than differences due to the choice of different demographic, mutation, or recombination parameters.
patterns of linkage disequilibrium
Here, we report a comparison of the average LD (as measured by r2) between the ARG-based simcoal2 program and fastsimcoal (SMC'-based) between two SNP markers located at a given recombination distance expressed in R units.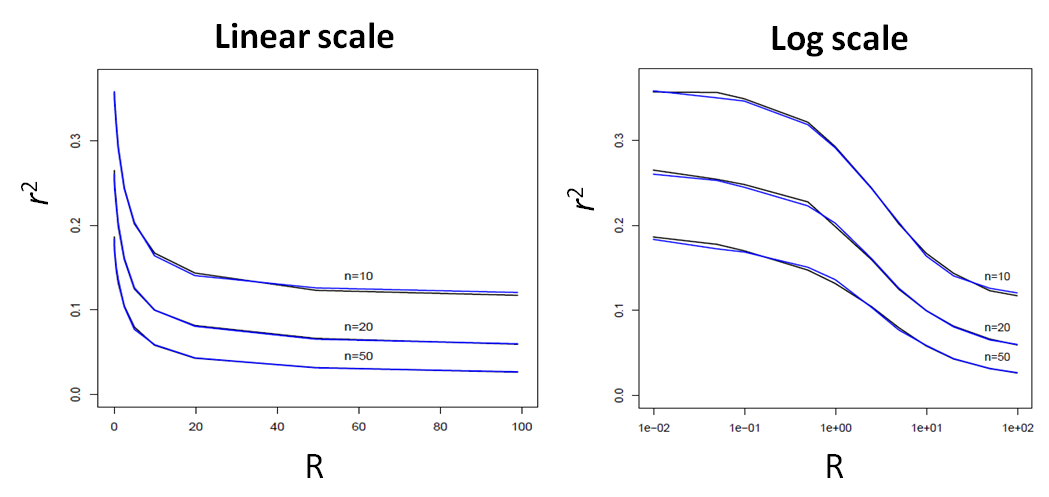

 |