Download latest release version 2.1.2executable filessource code |
|
We present here SIMCOAL2, an extended version of the SIMCOAL program (Excoffier et al. 2000), to simulate the neutral genetic diversity at partially linked loci under different histories and a wide range of migration and demographic models.
SIMCOAL2 includes a number of new features compared to the previous version:
The possibility of arbitrary recombination rates between adjacent loci
Multiple coalescent events per generation, allowing the correct simulation of very large samples and very large recombining genomic regions
The simulation of SNP data with arbitrary minimum frequency, for instance to simulate ascertainment bias
The output of diploid genotypic data generated under the assumption of Hardy-Weinberg equilibrium
The simulation of a mixture of different data types (DNA sequence, RFLP, STR, or SNP) along a single chromosome.
A recombination event can be schematically represented as follow:
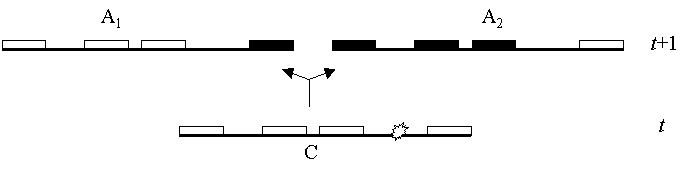
Schematic representation of a recombination event backward in time
Assuming that at
a given generation t we sampled 4 partially linked genes (loci)
arbitrarily distributed on a given chromosome segment C. A recombination event (represented
byon
the drawing) creates two new lineages in the ancestral generation, t+1,
here arbitrarily called A1 and A2. A1 receives from C
a copy of the genes located to the left of the recombination event, while A2
receives a copy of the genes located on the right side of the
recombination event. The copies of the
genes shown
in black are not observed in the present sample and thus have an
unknown origin. We make this distinction between known (white) and
unknown (black) genes, because
we only have to simulate the ancestry of the observed genes. Therefore,
we are only interested in the recombination
events occurring between white genes. The other recombination events
(e.g. between
the first two genes in A2) have no effect on the genetic constitution of the sample,
and thus do not need to be followed or simulated. The simulation of the
coalescent process with recombination is stopped when the Most Recent Common Ancestor
(MRCA) of each locus is found.
The
recombination rate is allowed to vary between adjacent loci in order to simulate
arbitrary patterns of recombinations along a chromosome, e.g. the occurrence of recombination hot-spots.
The following Figure gives an example of a simulated recombination
pattern. Here, four haplotypes blocks (noted as B1 to B4) characterized
by low recombination rate are interrupted by three hotspots of
recombination (HS1 to HS3). The height of the bars are here
proportional to the recombination rates between adjacent loci.
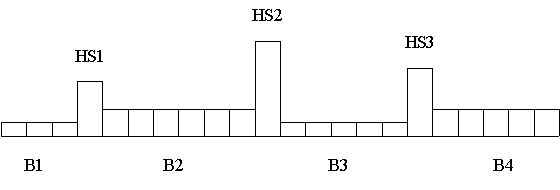
Representation of relative recombination rates across a chromosomal segment
Validation of the simulations: The expectation of the number of recombination events in the genealogy of two chromosomes studied for two partially linked markers was used to check the accuracy of our simulations. For two markers separated by the recombination fraction r, the expected number of recombination events having occurred since their Most Recent Common Ancestor is equal to 6R/(R+6), where R=4Nr (eq. 31 in Simonsen and Churchill 1997) and N is the population size. We report below the comparison between theoretical predictions and the average values obtained from the simulation of 10 000 genealogies with SIMCOAL2.
| R=1 | R=100 | |
| Expected | 0.86 | 5.66 |
| SIMCOAL2 | 0.87 | 5.67 |
The time to the MRCA
(TMRCA) at any locus (measured in units of M generations,
where M=N
for a haploid population, and M=2N
for a diploid population) is independent from the recombination rate. Under the
standard coalescent model where
only one coalescence per generation is assumed,
the expected
TMRCA
is given by 2(1-1/n), where n
is the number of sampled gene copies. When the sample size is not small with respect
to the effective size (Wakeley
2003), or when the recombination rate is high leading to the progressive
generation of a large number of ancestral chromosome segments (and thus
increasing the number of gene lineages in past generations), multiple coalescent
events needs to be implemented to avoid an overestimation of the
TMRCA
at any locus. Because this procedure is computationally intensive, we only
implement it if the effective number of gene lineages in a deme (at any given
time in the past) is large compared to the local deme size
N. On the
Figure below,
we present the
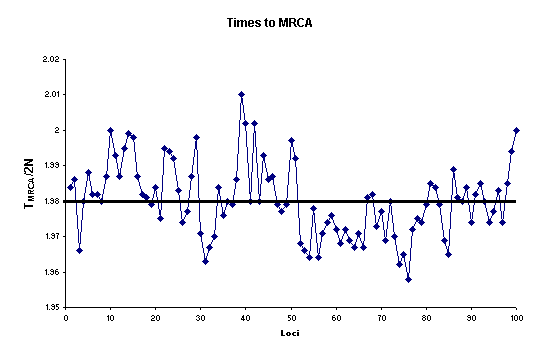
Distribution of TMRCA at 100 adjacent partially linked loci
With SIMCOAL ver 2.X users can generate the same data types as with SIMCOAL ver 1.0 (RFLP, DNA sequence, and microsatellite data), plus Single Nucleotide Polymorphims (SNP) data. We also offer the possibility to simulate SNP with a minimum frequency for the minor allele (which can be different for every SNP) in order to mimic one form of ascertainment bias. A SNP locus is modeled as a DNA base pair where one mutation has appeared somewhere at random along the genealogy of the locus. As shown in the following figure, the location of the mutation on the tree determines its frequency in the sample.
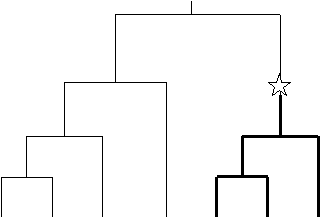
Schematic representation of a SNP-defining mutation on a gene genalogy
With the new release of SIMCOAL ver 2.1, it is now possible to simulate a much more flexible chromosomal structure than before. We now indeed offer the possibility to simulate the evolution of several chromosomes. By chromosomes we mean independent chromosomal segments. Users can simulate one or more (no limitation except hardware) chromosomes each one exhibiting its own number of blocks, its own number of markers, its own kind of markers etc... The input files used with SIMCOAL2.1 are thus different from those of SIMCOAL2.0 released earlier this year. The portions used to set up the populations features and their history are unchanged and the only differences are about the definition of the chromosomal segments. An example of a new input file is given below.
In SIMCOAL ver 2.1, we introduce a more realistic mutation model for STR markers: the Generalized Stepwise Mutation (GSM) model, which allows insertion (deletion) of more than one repeats. The number of repeats inserted (deleted) in one mutation event is treated as a random variable, and is randomly drawn from a Geometric distribution with parameter p. The value of p can be given in the input file (see below). The expectation and variance of the Geometric distribution are E[s]=p/(1-p) and Var[s]=p/(1-p)2, respectively, where s+1 is the number of mutational steps. Concretely this means that for a parameter p set up to 0 in the input file, the number of repeats inserted (deleted) will be equal to one for every mutation event. When p is set up to 0.5 in the input file, the expected number of repeats inserted (deleted) is equal to 2 with a variation around the mean is given in the following scheme.
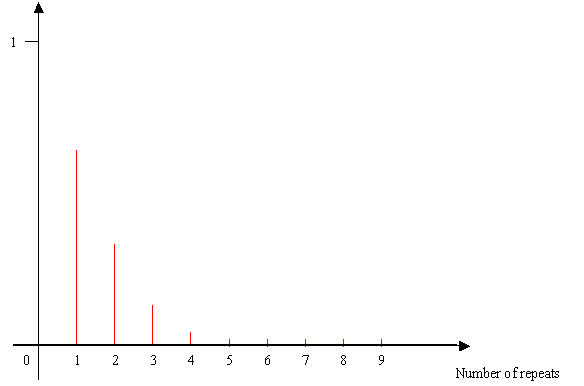
Distribution of the number of repeats inserted (deleted) according to a geometric law with parameter p=0.5
A single input file is needed for the simulation. The format of the input file is differs from the format used in SIMCOAL ver 1.0. The parameters required are listed bellow. The differences between the two versions of SIMCOAL are indicated using different colors. We show in blue the unmodified parameters, and in red the modified and new parameters.
| Demes sizes: | Demes can have different sizes. |
||||||||||||||||||||||||
| Samples sizes: | Sample sizes can also be unequal among
demes. Samples of size zero can also be used. |
||||||||||||||||||||||||
| Growth rate: | This is the exponential growth parameter
r in N(t) = N(0) exp(rt).
Negative growth implies population expansion, because we
simulate backward in time. |
||||||||||||||||||||||||
| No. of
migration matrices: |
Several matrices can be listed in the
input file. Their number is specified here. A value of zero means that
no migration matrix is provided, and the program will assume that
there is no migration between demes. |
||||||||||||||||||||||||
| Migration matrices: | A matrix of size equal to the number of
listed demes. If no matrix is defined, SIMCOAL creates a
migration matrix (with id "0") filled with
zeroes, which assumes no migration among demes. The matrices have
associated ids that correspond to their order of appearance in the
list, starting at zero. If no
particular migration matrix is specified with an
historical event at generation zero, the matrix
"0" (zero) is used. |
||||||||||||||||||||||||
| Historical events: | One first specifies the number of such
event to read. A value of zero is valid. Then, on separate lines, one enters each historical event in turn. They are structured as follows:
|
||||||||||||||||||||||||
| Number of independent chromosome segments : | This number refers to the number of
independent chromosome segments that needs to be simulated.
With the
release of SIMCOAL2 distributed before the
10 12 2004 it was not
possible to simulate different chromosomes with different features. In the
current release
of
SIMCOAL ver 2, it is now possible to simulate several chromosomes with
different kind and numbers of markers, different patterns of
recombination rates, different mutation rates (see the example given below). |
||||||||||||||||||||||||
| Number of blocks: | This number refers to the number of sets of partially linked loci (that we shall call blocks, hereafter) of the same data type, having the same mutation and recombination rates. For instance, to simulate L partially linked microsatellites with varying recombination rates and with different mutation rates, we need to specify L blocks, each one containing one microsatellite. To simulate a chromosome segment with L partially linked microsatellites uniformly spaced (with identical recombination rates between adjacent loci) and having identical mutation rates, one would only need to define a single block. | ||||||||||||||||||||||||
| Block specificities | For every block, we need to specify 3
parameters, as follows: 1) the
data type, 2) the number of loci to simulate and 3) the recombination
rate immediately on the right of each locus. Additionally to these 3 required parameters, several optional parameters need to be added depending on the data type. Optional parameters
* important note: The release of SIMCOAL 2.0 distributed before the 10.12.2004 does not support the GSM model for microsatellites, and does not accept the 5th parameter for microsatellites. Please update your version of SIMCOAL to ver 2.1 |
||||||||||||||||||||||||
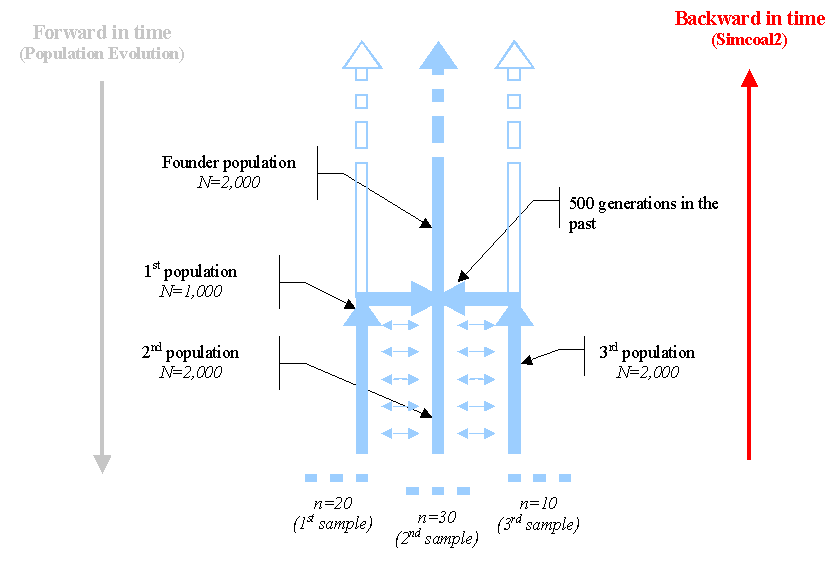
Definition of the history of population: Suppose that we want to simulate a scenario of a population trifurcation, where a (haploid) founder population of size N= 2 000 (note that it would be equivalent to the simulation of a diploid population of size N=1000) splits into 3 daughter populations of size 1,000, 2,000 and 1,000, respectively, where 20, 30 and 10 genes are respectively sampled. After the fission event, we shall assume that the three daughter populations are not completely isolated and can exchange genes at rate m=0.005 per generation, which is also assumed constant over time.
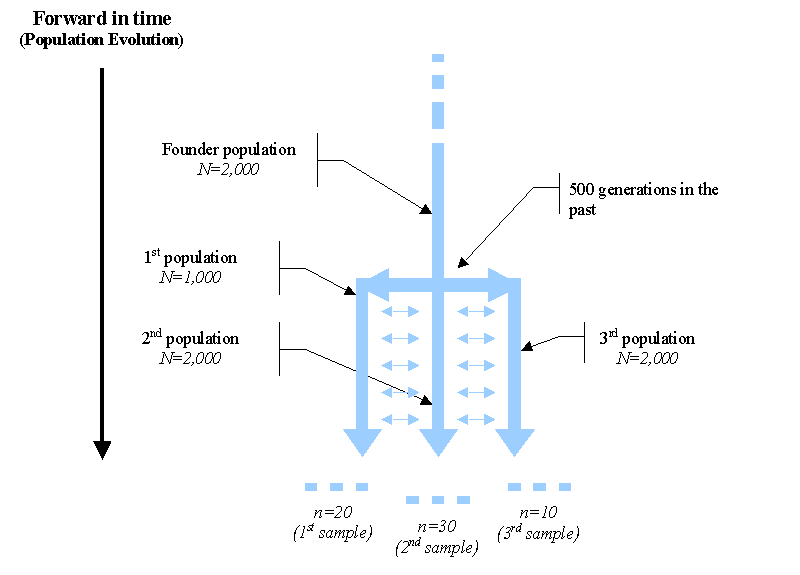
Schematic representation (viewed forward in time) of a population trifurcation
While the history of the populations is naturally described forward in time (black arrow on the left above), users simulating a coalescent process have to think backward in time as in the following Figure.
Schematic representation of the backward historical process actually simulated by SIMCOAL
The specification of the process to be simulated proceeds as follows:
//Input parameters for the coalescence and recombination simulation program : simcoal2.exe
3 samples to simulate
/Deme sizes (haploid number of genes)
1000
2000
1000
//Sample sizes
20
30
10
//Number of migration matrices : If 0 : No migration between demes
2
//Migration rates matrix 0 :
0.000 0.005 0.000
0.005 0.000 0.005
0.000 0.005 0.000
//Migration rates matrix 1 : No migrations
0 0 0
0 0 0
0 0 0
//Historical event: time, source, sink, proportion of migrants, new deme size, new growth rate, new migration matrix
2 historical events
500 0 1 1 1 0 1
500 2 1 1 1 0 1The first parameter (shown in red) represents the time at which occurs the historical event (in number of generations in the past relative to present time). The last parameter (shown in red as well) is the id of the new migration matrix. Thus, the first historical event states that: 500 generations ago, deme #0 (first population defined in the input file) will send all its genes to deme #1, and deme zero will remain at the same time as before, but a new migration matrix (#1) should be used from now on. the second historical event specifies that deme #2 should send all its genes to deme #1, and that migration matrix #1 should be used afterwards. Therefore at generation 501, all remaining gene lineages should be present in deme #1, and since no migrations are allowed, they will remain there until they all coalesce and the simulation stops.
Definition of the genetic structure of chromosomes: Suppose that we want to simulate the genetic diversity expected of a chromosomal segment made up of three blocks of partially linked loci (recall that by block we mean a set of identical genetic markers with the same inter-locus recombination rate), each one being separated by a recombination hot-spot, as shown in the following Figure. Assume that the first block contains 25 SNPs, that the second block contains 50 microsatellite (STR) loci, and that the last block is also made up of 25 SNPs.
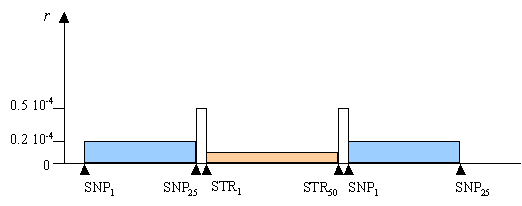
Recombination distribution to be simulated
If the blocks were each of consisting of 155 Kb, an assuming a diploid size of N=10,000, the recombination rate between adjacent loci (r) within the 3 blocks would be equal to 0.2 x 10-4, 0.1 x 10-4 and 0.2 x 10-4 , respectively. We shall finally assume that these blocks are separated by two recombination hot-spots of with recombinations rates r = 0.5 x 10-4.
Therefore, users will first have to set up the number of simulated blocks that they want to simulate. Here, this number is 5 because we need to include the two recombinations hot-spots. In SIMCOAL2, a block is defined as a set of adjacent markers with the same features and the same recombination rate between adjacent markers. A block can therefore be viewed as a chain of b elements. Every element has two attributes, which are the specificities of the genetic marker (see here to have more details on how to specificify the properties of the markers like its type, its mutation rate, etc... ) and the recombination rate with the next element. In the following scheme, the length of the links between markers is proportional to the recombination rate.
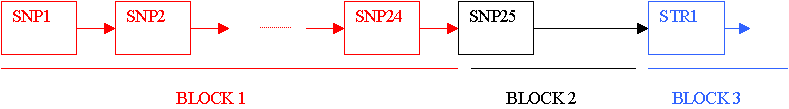
Note that the link between blocks i and i+1 will therefore depend on the recombination rate on the right of the last marker of block i. We therefore need to define a transition block (block2 in the previous scheme) made up of a single SNP with a large recombination rate in order to simulate the recombination hot-spot between the SNP#25 and the STR#1. In the input file these 3 blocks are defined as follows
| SNP | 24 | 0.00002 | 0.00000 | |
| SNP | 1 | 0.00005 | 0.00000 | |
| MICROSAT | 49 | 0.00001 | 0.00025 | 0 |
With these three blocks, we can simulates 25 SNPs with adjacent recombination rates equal to 0.00002, separated from 49 STR (with adjacent recombination rates equal to 0.00001 by a hot spot of recombination (recombination rates between SNP#25 and STR#1 equal to 0.00005). An identical structure would need to be described to simulate the second recombination hot-spot after the STR#50). Note that the last element of the last block is a special element, since its recombination rate is irrelevant, and ignored by SIMCOAL
With this kind of specifications, users are free to simulate any chromosomal structure they want, concerning the intensity of recombination rates but also the types of markers and the mutation rates.
For instance, if one would like to simulate 4 STRs with different mutation rates, with arbitrary recombination rates ( i.e. arbitrarily spaced on a chromosome), users would need to define 4 blocks of 1 STR as
| Kind of marker | Number of markers | Recombination rate | mutation rate | range constraint |
| MICROSAT | 1 | 0.00021 | 0.00250 | 0 |
| MICROSAT | 1 | 0.00041 | 0.00850 | 0 |
| MICROSAT | 1 | 0.00005 | 0.00742 | 0 |
| MICROSAT | 1 | 0.00000 | 0.00042 | 0 |
The following listing shows how to set up the complete input file to perform the simulations a trifurcating population described above.
|
============================%<====================================== //Input parameters for the coalescence and recombination simulation program : simcoal2.exe 3 samples to simulate //Deme sizes (haploid number of genes) 1000 2000 1000 //Sample sizes 20 30 10 //Growth rates 0 0 0 //Number of migration matrices : If 0 : No migration between demes 2 //Migration rates matrix 0 : 0.000 0.005 0.000 0.005 0.000 0.005 0.000 0.005 0.000 //Migration rates matrix 1 : No migrations 0 0 0 0 0 0 0 0 0 //Historical event: time, source, sink, proportion of migrants, new deme size, new growth rate, new migration matrix 2 historical events 500 0 1 1 1 0 1 500 2 1 1 1 0 1 //Number of independent (unlinked) chromosomes, and "chromosome structure" flag: 0 for identical structure across chromosomes, and 1 for different structures on different chromosomes. 4 1 //Number of contiguous linkage blocks in chromosome 1: 5 //Per Block: Data type, No. of loci, Recombination rate to the right-side locus, plus optional parameters ***see detailed explanation here*** SNP 24 0.00002 0.05 SNP 1 0.00005 0.10 MICROSAT 49 0.00001 0 0 0 MICROSAT 1 0.00005 0.0025 0 0 SNP 25 0.00002 0.10 //Number of contiguous linkage blocks in chromosome 2 2 //Per Block: Data type, No. of loci, Recombination rate to the right-side locus, plus optional parameters ***see detailed explanation here*** SNP 20 0.002 0.05 SNP 10 0.00002 0.10 //Number of contiguous linkage blocks in chromosome 3 1 //Per Block: Data type, No. of loci, Recombination rate to the right-side locus, plus optional parameters ***see detailed explanation here*** DNA 200 0.00002 0.00005 0.33 //Number of contiguous linkage blocks in chromosome 4 4 //Per Block: Data type, No. of loci, Recombination rate to the right-side locus, plus optional parameters ***see detailed explanation here*** SNP 20 0.002 0.05 MICROSAT 10 0.00002 0 0.5 10 MICROSAT 10 0.0005 0.0025 0 10 SNP 20 0.002 0.05 [EOF] ============================%<====================================== |
Summary of the different parameters needing to be defined for the description of the markers to be simulated
The parameters which can be given for every markers are:
MICROSAT [number of loci] [recombination rate] [mutation rate] [Geometric
parameter << NEW >>] [range
constraint]
SNP
[number of loci] [recombination rate] [minimum frequency]
DNA
[number of loci] [recombination rate] [mutation rate] [transition rate]
RFLP
[number of loci] [recombination rate] [mutation rate]
[number of
loci]
: Number of partially linked loci in a block
[recombination
rate]
: recombination rate between adjacent markers in a
block (Global recombination rate of the block = [recombination
rate] *([number of loci] -1))
[mutation
rate]
: mutation rate for every marker in a
block
(Global mutation rate of the
block = [mutation
rate] *[number of loci])
[Geometric parameter] :
parameter for the Geometric distribution (varies between 0 and 1)
[range
constraint]
: 0 or 1 means no constraints, higher numbers give the maximum number of
repeats
[minimum
frequency] :
minimum frequency of a SNP in the whole set of population (ascertainment
bias)
[transition
rate]
: fraction of substitutions that are transitions
Additional examples of input files for SIMCOAL (without recombination) can be found on this page .
The final state of each gene is recorded and output to several files according to two data formats. The first format is compatible with the Arlequin program (Schneider et al. 2000). The data generated after each simulation are output to a separate Arlequin project file. An Arlequin batch file is also created, listing all simulated files, and allowing one to compute different statistics on the whole set of simulated files with Arlequin. it therefore allows one to obtain their distribution after extraction from the Arlequin output files. Note that with version 2.0 of Arlequin, a utility is provided to collect most statistics computed in the analysis of batch files under the form of MS-Excel compatible files. Examples of statistics that can be extracted from batch file analysis by Arlequin are: nucleotide composition, gene diversity, homozygosity, mean number of pairwise differences, number of haplotypes, number of polymorphic sites, Tajima’s D, Fu's FS, extent of linkage disequilibrium among loci, mutation parameter q = 2Nu estimated from homozygosity, mean number of pairwise differences, number of polymorphic sites, or number of alleles, and genetic distances based on pairwise FST’s. Note also that the Arlequin software has a file conversion utility for exporting input data files into several other format like Biosys, Phylip, or GenePop, so that files produced by SIMCOAL2 could be also analyzed by these software's after file conversion.
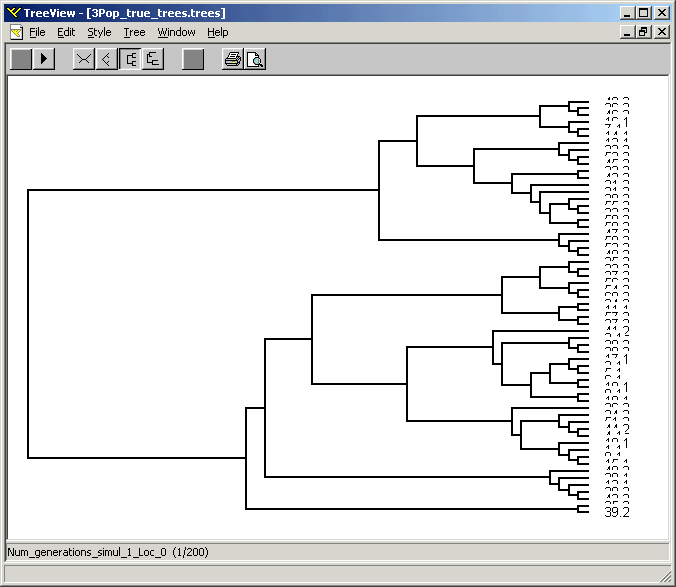
The two other sets of output file produced by SIMCOAL2 are compatible with the NEXUS file format. Two files with the “*.trees” extension list all the genealogical trees for each simulated locus. The branch lengths are expressed either 1) in units of generations scaled by the population size (N), and therefore representing the true coalescent history of the sample of genes, or 2) in units of average number of substitutions per site (except for data sets containing SNP markers), and therefore representing the realized mutational tree. These two sets of trees can be visualized with the software TREEVIEW (Page 1996), as shown on the figure below. Finally, a file with the “*.paup” extension, lists all the simulated genes together with their true genealogical structure. This file can be analyzed with David Swofford's PAUP* software (1999). Because, these files can be huge, we do not produce them by default, and a conditional compilation directives (#define _PRINT_TREE_PAUP_ and ) must be uncommented in the source code of "cond_var.h", and the source must be recompiled to reflect these changes.
Unlike SIMCOAL ver 1, SIMCOAL ver 2 stores all the output files in a folder automatically created and named according to the name of the input parameter file (without the extension .par). Beware, SIMCOAL2 does not clean your disk. For example: if you run a first set of 1000 simulations and a second set of 100 simulations using the same input parameter file, only the first 100 Arlequin (*.arp) files will be updated. The others will be kept on your disk without any changes.
The speed of simulation with recombination can be much slower than without recombination, and will increase with sample size, effective population size, the number of loci, and the total recombination rate. For instance, SIMCOAL2 needs 15 minutes to simulate 10,000 samples of 100 diploid individuals (n=200 and N=1,000) typed at 250 SNPs over a segment of 250 Kb (assuming 1cM = 1Mb, and thus with total recombination rate R=4Nr=10 for the whole chromosome segment), and about 2.5 hours to simulate 10,000 samples of 250 microsatellite loci over 2.5 Mb (total recombination rate R=100), using a Pentium 4 (2.8Ghz) under the Linux operating system.
The archive of executable files for windows and Linux includes the compiled files simcoal2.exe as well as examples of input files to simulate partially linked SNPs, microsatellites and a mixture of these data types. After extraction, there are three ways to run simcoal:
The source files can be compiled with Borland C++ Builder v. 5 and v. 6 to run SIMCOAL2 on windows (2000/XP), or with gcc (GNU project) to run SIMCOAL2 on a Linux Operating System. To compile the source code under Linux, uncomment the line "#define _LINUX_" in the file "cond_var.h". Others compilation options are also listed in the file "cond_var.h", and enable SIMCOAL2 to produce additional (undocumented) output files, mainly used for debugging purposes.
Wakeley J, and Takahashi T, 2003. Gene genealogies when the sample size exceeds the effective size of the population. Mol. Biol. Evol. 20(2):208-213.
Guillaume Laval, Computational and Molecular Population Genetics Lab, University of Bern
Last edited on 14.09.05 (16:14)